
Posts
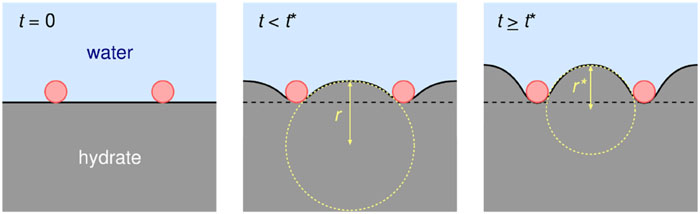
Statistical mechanical theory and computational study on thermodynamic stability of clathrate hydrates
A new paper from our group has been published. Clathrate hydrates are non-stoichiometric inclusion compounds with critical relevance to energy resources and CO2 sequestration, formed by guest molecules encapsulated in water cages. This perspective overviews the synergistic progress achieved through statistical mechanics and molecular simulation with intermolecular potential models in three key areas: thermodynamic stability, structural polymorphism, and dynamic processes. Theoretical estimation of its stability, originated from the van der Waals and Platteeuw theory, has been greatly improved by revisions accounting for constant pressure conditions, multiple occupancy, and host–guest coupling, enabling accurate prediction of multi-phase coexistence.
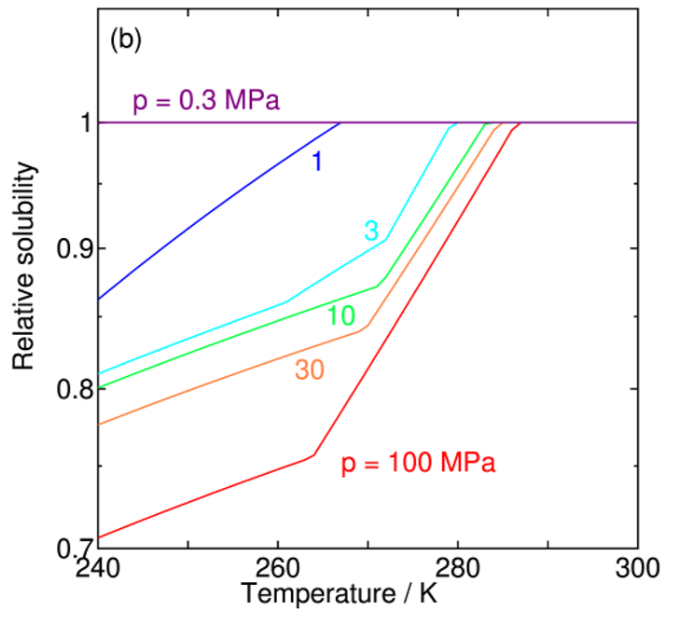
Solubility of Water in Carbon Dioxide
A new paper from our group has been published. This paper investigates the solubility of water in liquid carbon dioxide (CO2) under conditions where water or CO2 hydrate (clathrate hydrate) coexists, using theoretical calculations. The main focus is to quantitatively evaluate the reduction in solubility caused by hydrate formation under low-temperature and high-pressure conditions, and to clarify its temperature and pressure dependencies. This research provides valuable insights into thermodynamic properties related to practical challenges in large-scale CO2 transport, such as pipeline blockage and corrosion in carbon capture and storage (CCS).
Ice T2 has been synthesized! (University of Tokyo)
More than 20 different crystal structures of ice have been discovered across various temperature and pressure ranges. This is considered an unusually large number of polymorphs for a pure substance. Of these, 12 were discovered by the 20th century, and the remaining 8 have been found since the 21st century, with the pace of discovery remarkably accelerating. Following the synthesis of plastic ice reported at the beginning of this year, there has been a report of the successful synthesis of ice T2, which our group predicted in 2018.
Plastic ice has been synthesized! (University of Rome)
The existence of plastic ice (plastic crystalline ice), predicted by the Tanaka group (the predecessor of our current laboratory) in 2008, has apparently been confirmed. Ice VII, which forms under ultra-high pressure, is said to be extremely hard. However, when heated, before the crystal structure collapses entirely, it transforms into a very soft crystal (barely maintaining its structural integrity) where water molecules can rotate freely. This state is called plastic ice (plastic crystalline ice).
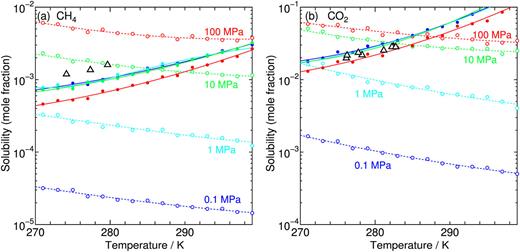
On the phase behaviors of CH4–CO2 binary clathrate hydrates`:` Equilibrium with aqueous phase
A new paper from our research group has been published. We explore the solubilities of guest CH4 and/or CO2 in the aqueous state coexisting with the corresponding hydrate. The equilibrium conditions are estimated by calculating the chemical potentials of water and guest species in the hydrate on the basis of a statistical mechanical theory using pairwise intermolecular potentials. This requires the least computational cost while covering a wide range of temperature, pressure, and composition of guest species, even for the binary hydrate.
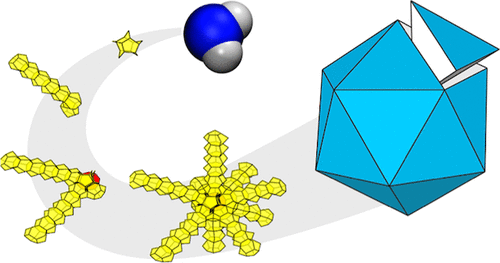
Emergence of the pentagonal ice nanocrystal
A new paper from our research group has been published. Press Release (in Japanese) Multitwinned nanocrystals are commonly found in substances that preferentially adopt tetrahedral local arrangements, but not yet in water crystals. Ice nanocrystals are pivotal in cloud microphysics, and their surfaces become increasingly prominent in determining structure as crystal size decreases. Nevertheless, discussions on nanocrystal structures have predominantly centered on ice polymorphs observed in bulk: hexagonal (Ih), cubic (Ic), and stacking-disordered (Isd) ices.
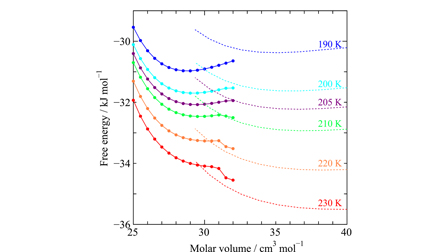
Stability mechanism of crystalline CO2 and Xe
A new paper from our research group has been published. We explore the phase behaviors of simple molecular crystals in order to investigate the molecular basis of the stability mechanism relative to their liquid counterparts. The free energies of the face centered cubic crystals of Xe and CO2 are calculated as a collection of oscillators, and those of the liquids are from an equation of state via molecular dynamics simulations. The vibrational free energy in the solid is separated into the harmonic and anharmonic terms.
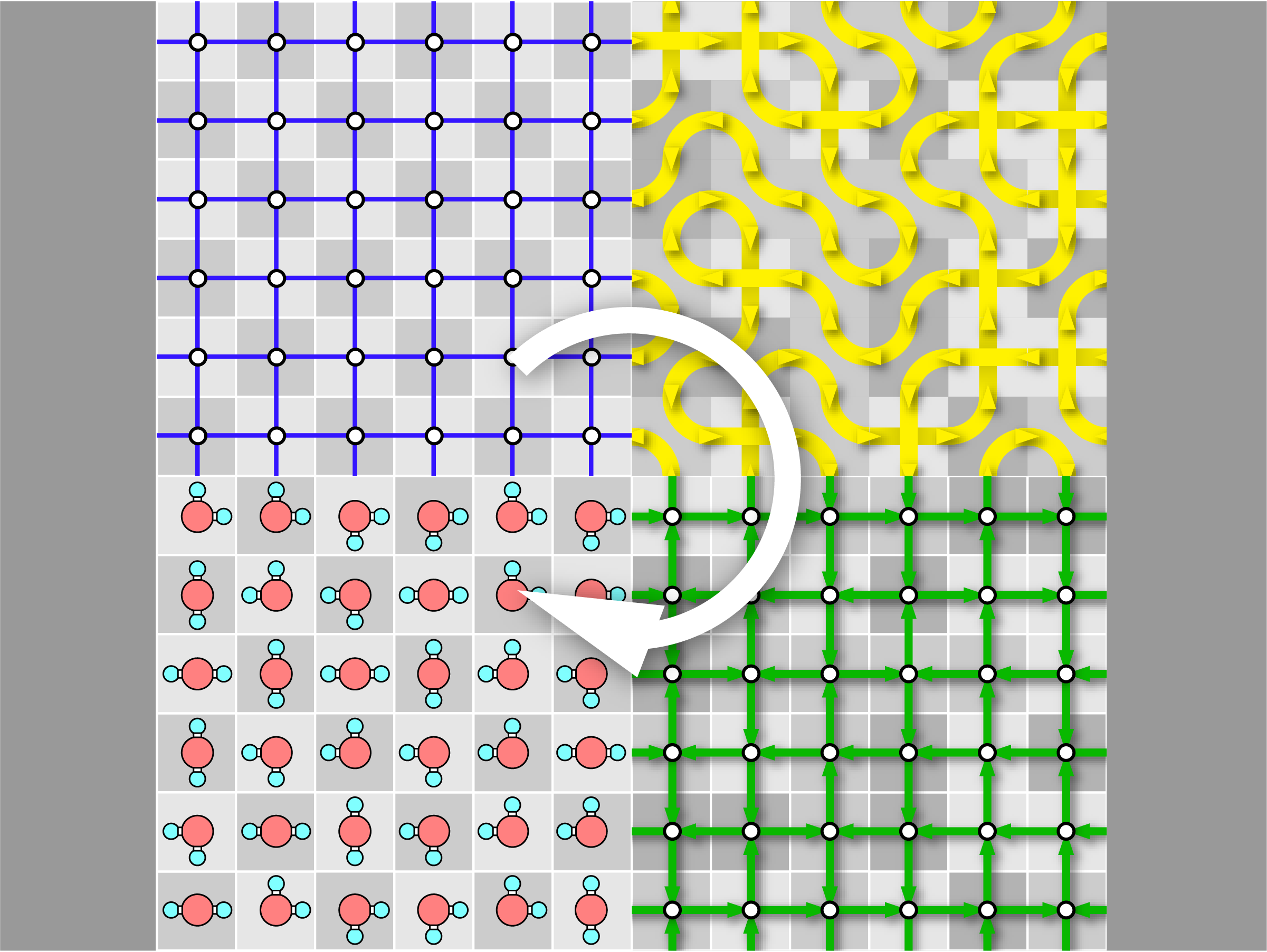
GenIce-core:Efficient Algorithm for Generation of Hydrogen-Disordered Ice Structures
A new paper from our research group has been published. Ice is different from ordinary crystals because it contains randomness, which means that statistical treatment based on ensemble averaging is essential. Ice structures are constrained by topological rules known as the ice rules, which give them unique anomalous properties. These properties become more apparent when the system size is large. For this reason, there is a need to produce a large number of sufficiently large crystals that are homogeneously random and satisfy the ice rules.
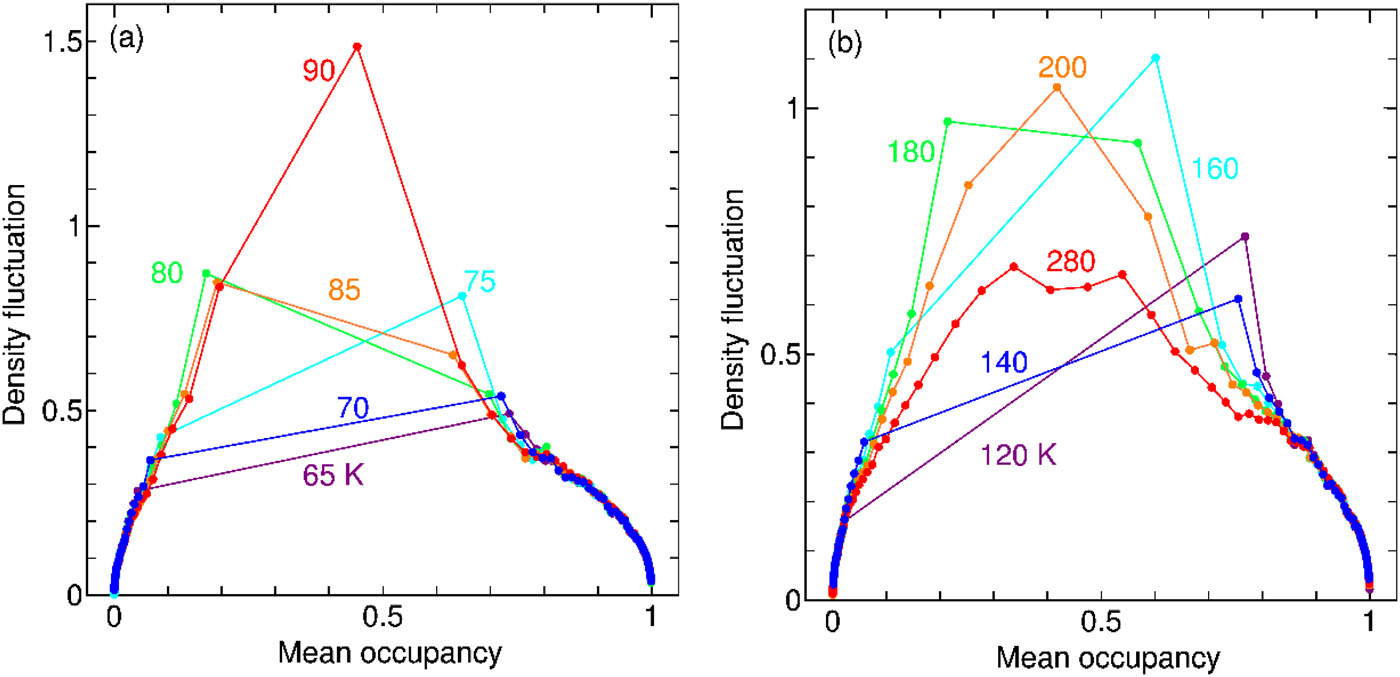
Cage occupancies of CH4, CO2, and Xe hydrates:Mean field theory and grandcanonical Monte Carlo simulations
A new paper from our research group has been published. We propose a statistical mechanical theory for the thermodynamic stability of clathrate hydrates, considering the influence of the guest–guest interaction on the occupancies of the cages. A mean field approximation is developed to examine the magnitude of the influence. Our new method works remarkably well, which is manifested by two sorts of grandcanonical Monte Carlo (GCMC) simulations. One is full GCMC, and the other is designed in the present study for clathrate hydrates, called lattice-GCMC, in which each guest can be adsorbed at one of the centers of the cage.
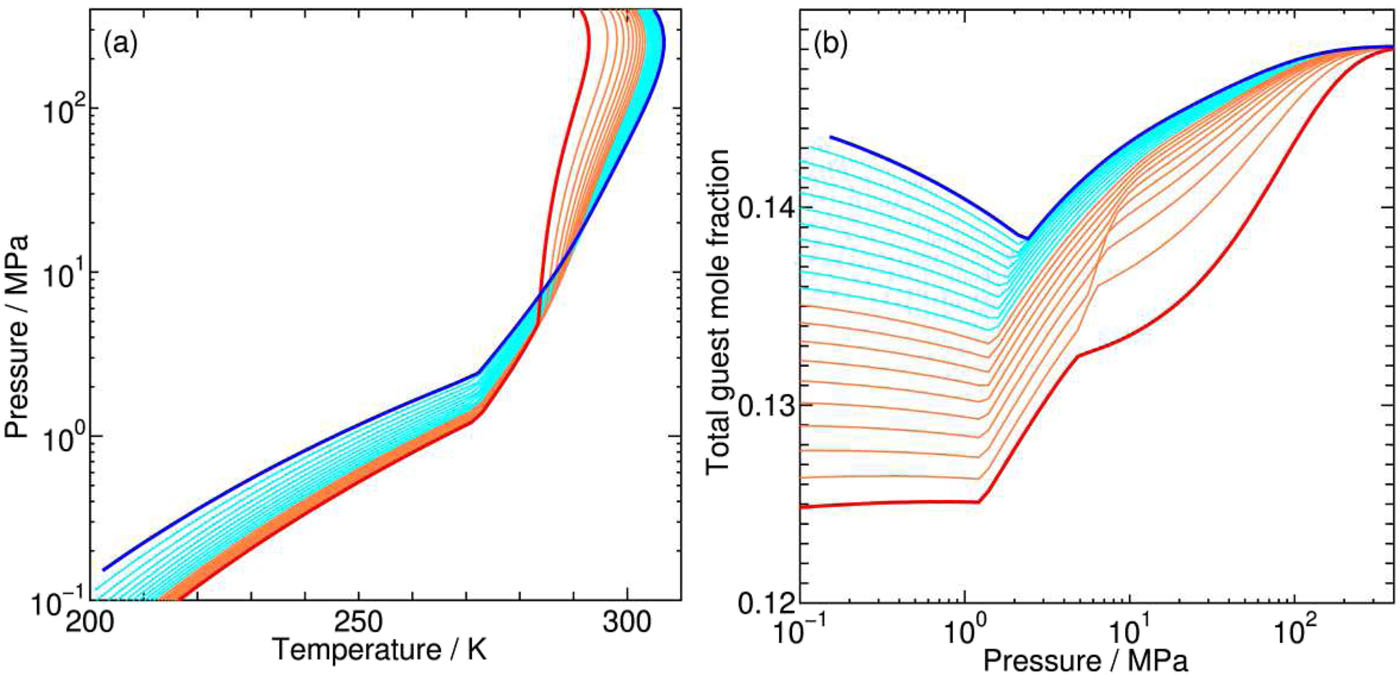
Efficiency and energy balance for substitution of CH4 in clathrate hydrates with CO2 under multiple-phase coexisting conditions
Many experimental and theoretical studies on CH4–CO2 hydrates have been performed aiming at the extraction of CH4 as a relatively clean energy resource and concurrent sequestration of CO2. However, vague or insufficient characterization of the environmental conditions prevents us from a comprehensive understanding of even equilibrium properties of CH4–CO2 hydrates for this substitution. We propose possible reaction schemes for the substitution, paying special attention to the coexisting phases, the aqueous and/or the fluid, where CO2 is supplied from and CH4 is transferred to.
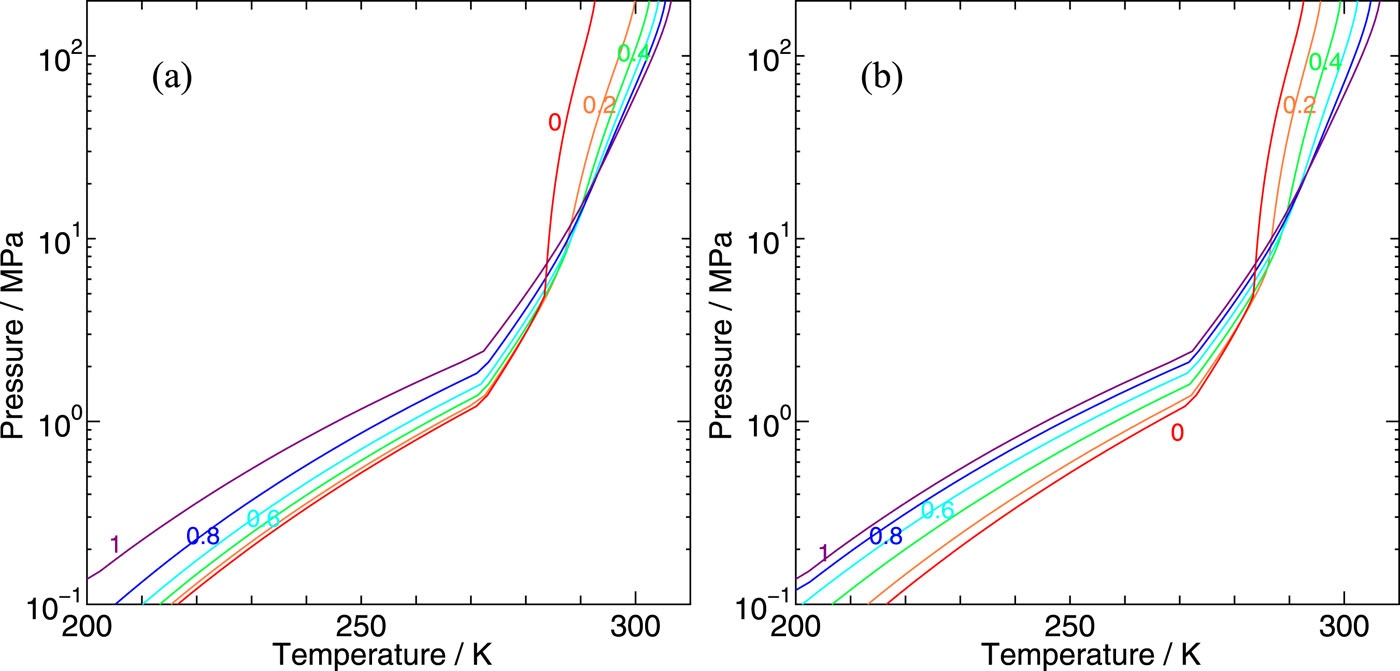
On the phase behaviors of CH4–CO2 binary clathrate hydrates:Two-phase and three-phase coexistences
We develop a statistical mechanical theory on clathrate hydrates in order to explore the phase behaviors of clathrate hydrates containing two kinds of guest species and apply it to CH4–CO2 binary hydrates. The two boundaries separating water and hydrate and hydrate and guest fluid mixtures are estimated, which are extended to the lower temperature and the higher pressure region far distant from the three-phase coexisting conditions. The chemical potentials of individual guest components can be calculated from free energies of cage occupations, which are available from intermolecular interactions between host water and guest molecules.
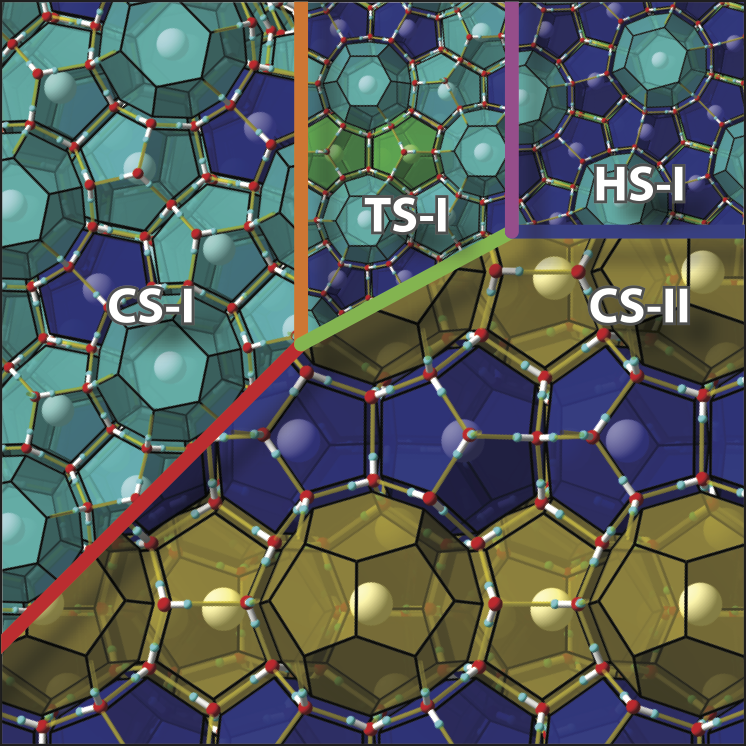
Structure Selectivity of Mixed Gas Hydrates and Group 14 Clathrates
A new paper from our group has been published. In a previous paper, we examined the regularity with which the crystal structure of inclusion hydrates is chosen. We applied that approach to a new mixed gas inclusion hydrates and group 14 clathrate compounds. In the former, we presented an overarching explanation for why mixing gases may change the crystal structure. Adding just a few molecules of a certain type may significantly change the crystal structure.

On the role of intermolecular vibrational motions for ice polymorphs. III. Mode characteristics associated with negative thermal expansion.
It is well known as an unusual property of liquid water that when it is cooled down, it begins to expand at a temperature below 4 degree celsius. When it is cooled down to 0 degree, it becomes ice, and after that, its volume becomes smaller as it is cooled down. However, even after it becomes ice, if the temperature is kept very low, it begins to expand again at an absolute temperature of 60 K or lower.
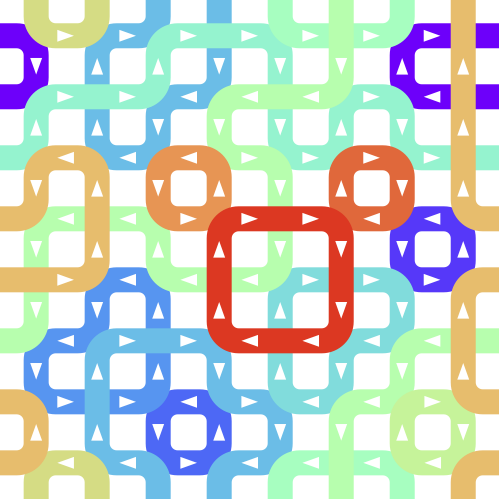
On the anomalous homogeneity of hydrogen-disordered ice and its origin
In hydrogen-disordered ice, each water molecule is oriented in a different way, and the interaction between one water molecule and the surrounding water molecules can be attractive (low interaction energy) or repulsive (high interaction energy). The interaction between a water molecule and a large number of surrounding water molecules seems to be most influenced by the orientation of the water molecules in the immediate vicinity. However, calculations and experiments have shown that the interaction between a water molecule and all surrounding water molecules is almost the same regardless of whether the direction of the nearby water molecule is attractive or repulsive.
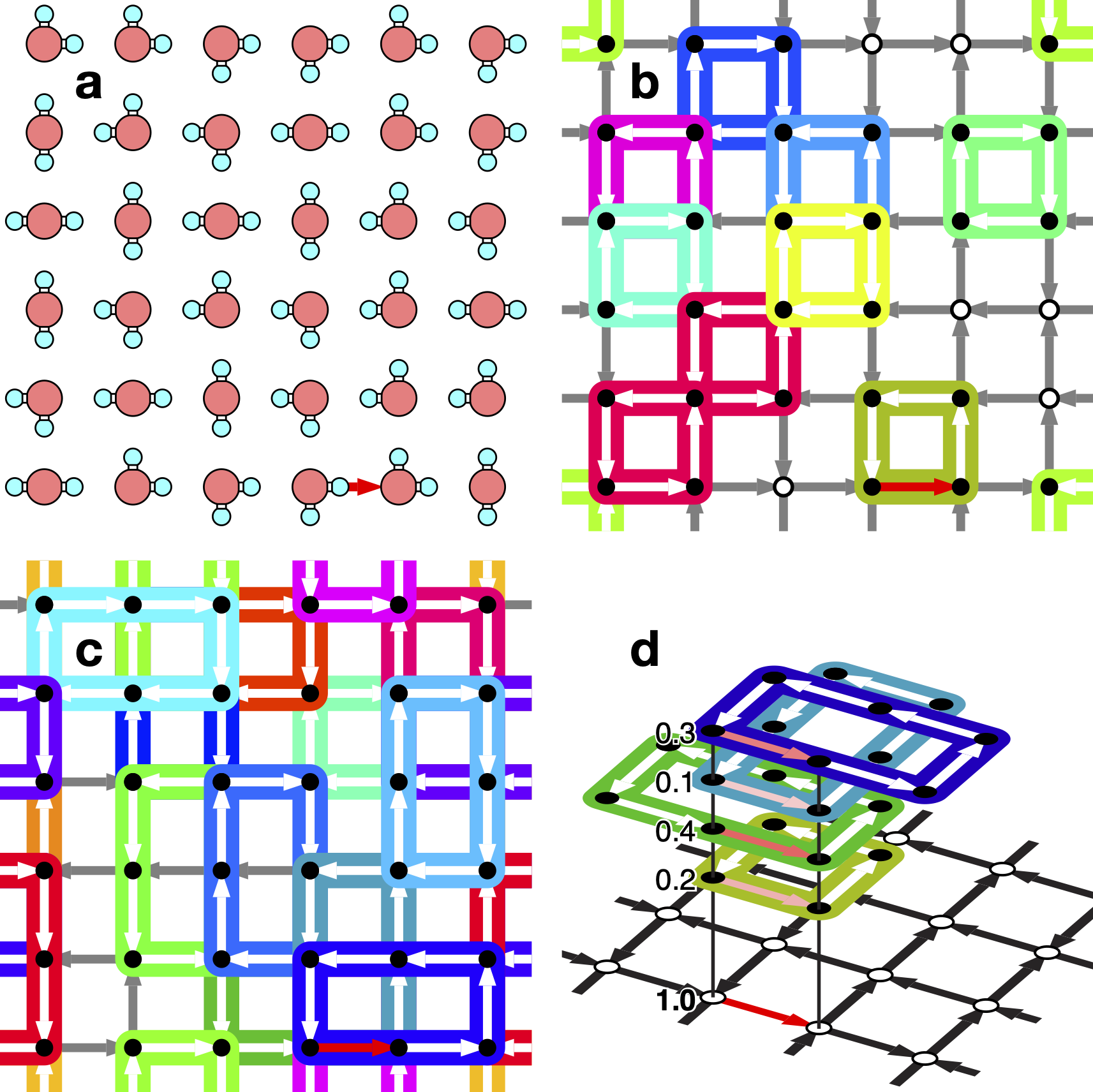
Novel Algorithm to Generate Hydrogen-Disordered Ice Structures
A new paper from our group has been published. There is a growing demand for computer simulations of ice, which is composed of a large number of molecules, to study the behavior of molecules dissolved in small amounts in ice and to search for new crystal structures. Unlike ordinary crystals, the water molecules in ice crystals are not oriented in the same way. In order to handle ice in computer simulations, it is necessary to generate crystal structures with randomized molecular orientations in an appropriate manner.
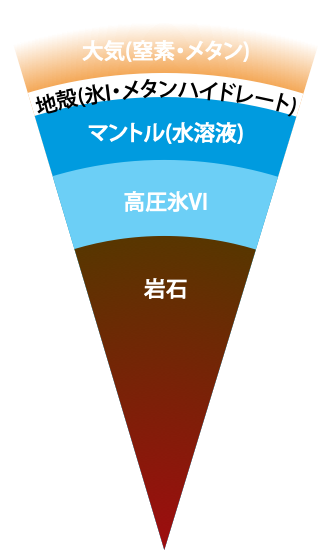
On the Occurrence of Clathrate Hydrates in Extreme Conditions:\ Dissociation Pressures and Occupancies at Cryogenic Temperatures with Application to Planetary Systems
We investigate the thermodynamic stability of clathrate hydrates at cryogenic temperatures from the 0 K limit to 200 K in a wide range of pressures, covering the thermodynamic conditions of interstellar space and the surface of the hydrosphere in satellites. Our evaluation of the phase behaviors is performed by setting up quantum partition functions with variable pressures on the basis of a rigorous statistical mechanics theory that requires only the intermolecular interactions as input.
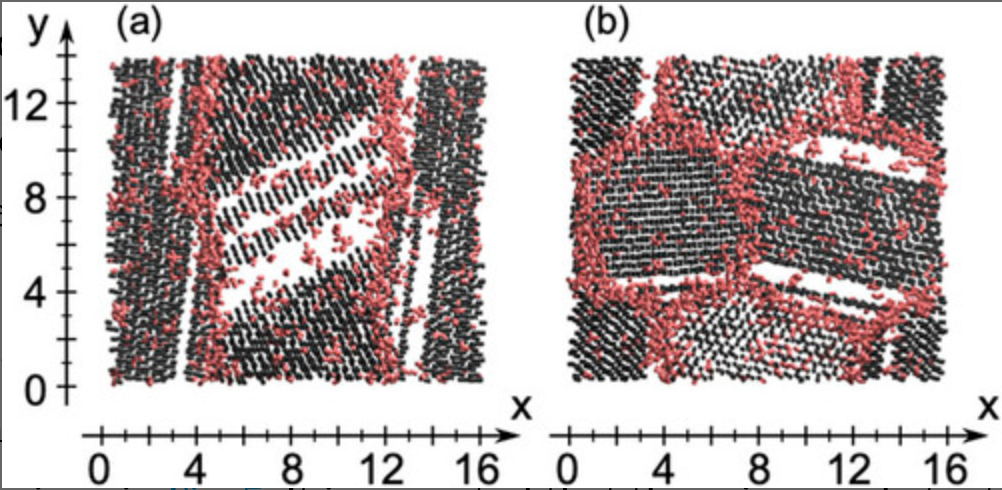
Molecular dynamics study of grain boundaries and triple junctions in ice
We perform classical molecular dynamics simulations of polycrystalline ice at 250 K using the TIP4P/Ice model. The structures of polycrystalline ice are prepared by growing ice particles in supercooled water. An order parameter developed recently is used to characterize local structures in terms of the liquid–liquid phase transition scenario. It is shown that the grain boundaries and triple junctions in ice are structurally similar to low-density liquid water in which most water molecules form four hydrogen bonds and the O–O–O angles deviate from the tetrahedral angle of 109.
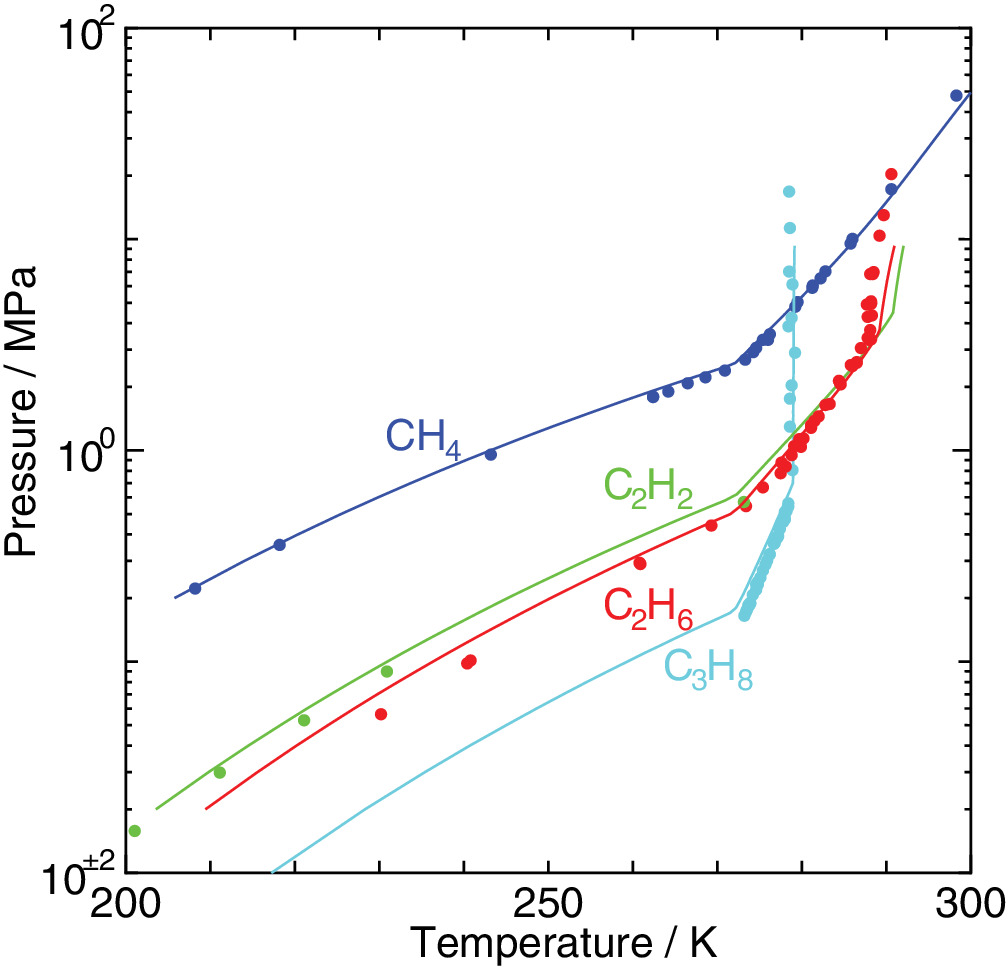
Cage occupancy and dissociation enthalpy of hydrocarbon hydrates
An elaborated statistical mechanical theory on clathrate hydrates is applied to exploration of their phase equilibria and dissociation enthalpies. The experimental dissociation pressures of methane, ethane, acetylene, and propane hydrates are well recovered by the method we have proposed. We estimate water/hydrate and hydrate/guest two‐phase coexisting conditions in the temperature, pressure, and composition space in addition to three‐phase equilibrium conditions. It is shown that the occupancy of guest molecules and the two‐phase boundaries in the phase diagram vary depending sensitively on its size.
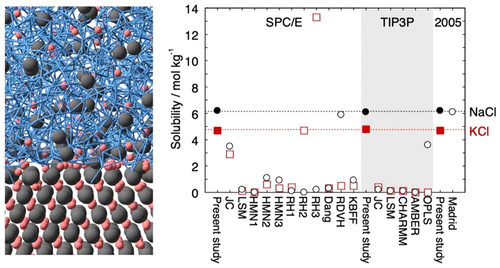
Lennard-Jones Parameters Determined to Reproduce the Solubility of ions
Most classical nonpolarizable ion potential models underestimate the solubility values of NaCl and KCl in water significantly. We determine Lennard-Jones parameters of Na+, K+, and Cl- that reproduce the solubility as well as the hydration free energy in dilute aqueous solutions for three water potential models, SPC/E, TIP3P, and TIP4P/2005. The ion–oxygen distance in the solution and the cation–anion distance in salt are also considered in the parametrization. In addition to the target properties, the hydration enthalpy, hydration entropy, self-diffusion coefficient, coordination number, lattice energy, enthalpy of solution, density, viscosity, and number of contact ion pairs are calculated for comparison with 17 frequently used or recently developed ion potential models.
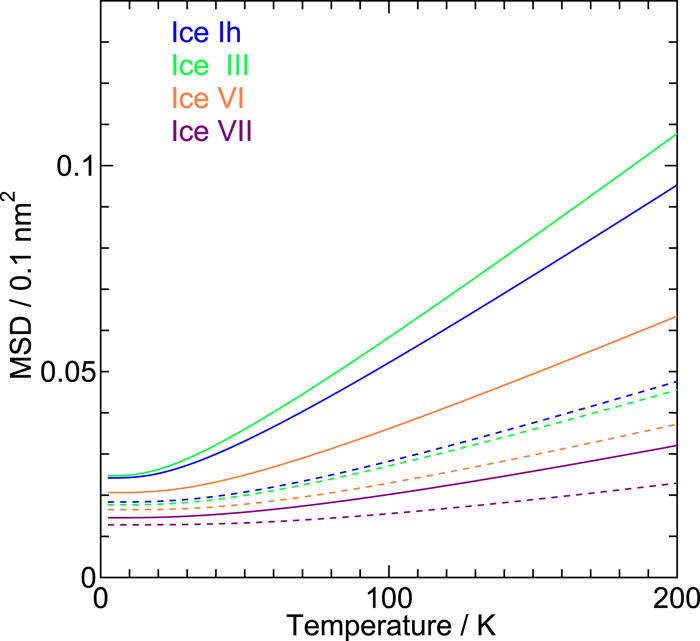
On the role of intermolecular vibrational motions for ice polymorphs II:\ Atomic vibrational amplitudes and localization of phonons in ordered and disordered ices
We investigate the vibrational amplitudes and the degree of the phonon localization in 19 ice forms, both crystalline and amorphous, by a quasi-harmonic approximation with a reliable classical intermolecular interaction model for water. The amplitude in the low pressure ices increases with compression, while the opposite trend is observed in the medium and high pressure ices. The amplitude of the oxygen atom does not differ from that of hydrogen in low pressure ices apart from the contribution from the zero-point vibrations.